
3D Printed Medical Devices – Insight into the FDA Guidance on Additive Manufacturing
Additive Manufacturing (AM) or 3D printing – offers new and exciting ways to integrate parts not only for prototypes, but for final products. It presents novel ways of thinking about design, as new rules govern how parts are made.
The FDA has stepped into the space of AM by providing a non-binding technical guidance to medical device designers and manufacturers. As with devices made using traditional methods, devices made with AM are subject to regulatory requirements based on how they are used, which drive their class and subsequent submission requirements.
Additive Manufacturing components are created using a layer-by-layer additive method, with the additive mechanism and material types varying widely. Traditionally, subtractive methods (e.g. lathe and mill) or net shape processes (where parts need to be designed to be removed from a tool) were the only choices available to fabricate components in a device. Traditional methods of fabrication require a tool to remove material – with AM this is no longer needed, as layers are built up by tracing a 2D profile at each layer cross section. AM methods provide technical advantages, such as the ability to build internal geometries during fabrication and easily integrate several materials into one part. The advantages provided by AM, however, require additional considerations when used in medical device applications.
Expanded Parameters
New challenges arise with Additive Manufacturing that give cause for consideration, especially when controls are required for processes used in medical device development. AM technologies not only require the input and control of traditional parameters – such as feed rate or cure time – they also need a new list of settings. It is key to document these parameters with tolerances and justification, ensuring that they are monitored and locked during fabrication. The parameters for AM processes include:
- Build orientation
- Build/cure pathway
- Layer thickness (z-axis resolution)
- Layer thickness (x/y-axis resolution)
- Cure speed/melt flow rate
- Cure/melt power level
- Curing beam focal point
- Melt nozzle diameter
- Build chamber ambient temperature
- Build platform temperature
- File conversion resolution
- Input material (virgin vs. reused)
- Fill density
- Fill pattern
- Shell thickness
- Build material properties, including
- Identification (CAS number, material standard, chemical name)
- Supplier
- Specifications
- Certificates of analysis (COAs), including test methods (e.g. ISO or ASTM)
Varying the above parameters can lead to a variety of effects which can affect the resulting part(s), including changes in:
- Material porosity
- Part exterior finish
- Entrapped virgin material
- Molecular structure of build material
These effects must be identified in documentation with justification for their presence.
Additive Manufacturing and the FDA
The FDA’s technical guidance gives an overview based on a workshop undertaken between the FDA and stakeholders from industry (including medical device manufacturers, AM companies, and academia). The intention of the guidance is to provide an overview and obtain feedback from broader industry and experts in the field as to the direction for regulation of medical devices produced via AM.
The workshop focused on five broad themes and four common processes:
Themes
- Materials
- This included plastics, metals, and ceramics
- This did not include biological AM materials (e.g. tissue or cells)
- Design, printing, and post-printing validation
- Printing characteristics and parameters
- Physical and mechanical assessment of final devices
- Biological considerations of final devices (e.g. cleaning, sterility, biocompatibility)
Processes
- Fused filament fabrication (FFF/FDM)
- Powder bed fusion (PBF/SLS)
- Stereolithography (SLA)
- Liquid-based extrusion (LBE)
The key recommendation in the guidance is that a given AM process be validated thoroughly using these themes. Ensuring that process parameters are verified and tracked, fabrication can be locked down to known critical steps. There is an obligation for AM manufacturers to document and control the progression from input materials to output parts, ensuring that process steps haven’t altered the components outside of specifications.
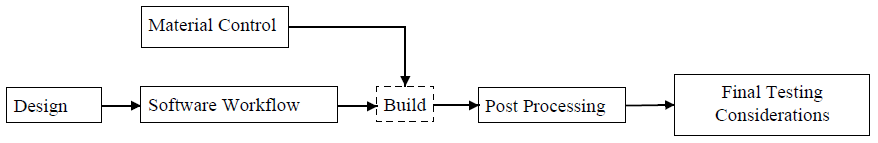
AM deals with a variety of input materials in granular or liquid form (process dependant), and final materials should be tested at given parameters to ensure consistency across volume builds. This will ensure that curing power levels don’t alter material chemistry through overheating. Also, many vendors are driven to reuse material in processes – while this reduces waste (both monetary and environmentally), it can lead to repeated heating cycles causing unexpected material chemistry and mechanical properties. Methods of excess material removal vary across many AM processes, and the FDA recommends that these processes be described in detail. Any methods to remove or justifications to retain excess uncured material on or within components should be documented in consideration of safety and effectiveness of the component per its intended use.
Part dimensions and aspects can shift based on their location on the build platform, including variations amongst machines of the same type. The guidance recommends that individual machines are validated before use to quantify these changes. The scope of validation includes both OQ (Operation Qualification) and IQ (Installation Qualification) of a given AM machine. Furthermore, ongoing and continuous validation testing can mitigate risks introduced by these and other factors, such a drift away from a calibrated base point over time.
The ability to create complex internal geometries and varying porosities with AM is one of its most appealing technical advantages; however, the guidance highlights these geometries and density adjustments can be concerning in biocompatibility and sterility requirements. This is due to the potential for increased contact surface area and regions that could resist certain sterilization methods. Due to the lack of cleaning or flushing methods available for internal portions of parts, the cleanliness of the build material and environment is crucial. Also, considerations for cleaning the patient-contacting surfaces must be considered not only directly after fabrication but upon reuse of a device. Information is needed to demonstrate that the device is cleaned before being provided to the end user, with this information included in a premarket submission. Reusable devices should include reprocessing instructions (for cleaning and sterilization) in the device’s labeling. Furthermore, surface finish requirements should be outlined in the product specifications.
Biocompatibility requirements remain the same as other medical devices, with ISO-10993-1 as the standard metric used for testing. If any chemical additives with known toxicities are used, more information may be necessary in a submittal as defined by ISO-10993-1.
Changing parameters with a validated AM machine, such as its location, external environmental temperature, material, or orientation requires a revalidation of the system with documentation. Maintaining known settings for output parts creates necessary consistency.

Summary
Overall, the FDA is approaching 3D Printing/Additive Manufacturing (AM) for medical devices using previously established processes for medical devices. Additional technical considerations (see both Themes and Processes above) due to AM technologies must be considered in submissions. While engaging with experts in the field, the FDA has highlighted key parameters and their documentation of these to ensure that processes can create consistent components for medical device use. Validation of the process, as outlined by Figure 1, is emphasized.
As Additive Manufacturing technologies expand and their applications grow, the FDA will continue to evaluate submissions for new devices to determine safety and effectiveness, and it will be essential to keep up with these updates in the days to come.
For more information, find the full published guidance published on the FDA’s website.
Nathan Müller, EIT, is a StarFish Medical Mechanical Engineer. He is passionate about the applications of biotechnology and biomedical science in product development, with core competencies in biomedical-mechanical design and material sciences.
Photo source: fda.gov/