
FDA Guidance on Computational Modeling and Simulation in Medical Device Submissions
The FDA’s Modeling & Simulation (M&S) Working Group of the Senior Science Council has noted that the use of Computational Modeling and Simulation (CM&S) is expected to increase in the future years, in a report outlining the Successes and Opportunities in Modeling & Simulation for FDA. FDA has recognized the importance of using CM&S in medical device development.
This article summarizes the FDA Guidance Document “Assessing the Credibility of Computational Modeling and Simulation in Medical Device Submissions (17th November 2023)”.
Computational modeling is the use of computers to simulate and study complex systems using mathematics, physics, and computer science. Simulation is achieved by adjusting the variables independently, or in combination with each other, and observing the outcomes. A StarFish Medical whitepaper, Computational Modeling and Simulation (CM&S) in Medical Devices, provides a broad overview of CM&S and its usefulness in the medical device industry.
CM&S can greatly improve the product development lifecycle by providing a cheaper and faster pathway to arrive at the final product design. Medical Device companies can also execute and collect evidence of device safety and performance without the need to develop multiple Proofs of Concepts (POCs) for assessment.
FDA has issued a few guidances and reports around CM&S, identifying that computational modeling and simulation studies can be used to add value to bench testing and nonclinical in vivo and clinical studies used to evaluate the safety and effectiveness of medical devices.
The CM&S Medical Device Submission FDA Guidance Document “Assessing the Credibility of Computational Modeling and Simulation in Medical Device Submissions” provides a general risk-informed framework to assess the CM&S during regulatory submissions. The guidance is heavily based on the FDA recognized standard ASME V&V 40 Assessing Credibility of Computational Modeling through Verification and Validation: Application to Medical Devices, a standard that identifies a framework for assessing verification, validation, and uncertainty quantification (VVUQ).
The guidance provides a nine-step process for developing and assessing the credibility of CM&S for regulatory submissions.
Define how the model will be used and perform a risk assessment.
The first three steps of the framework involve defining the way the computational model will be used and the risk of using the model.
- Describe the Question(s) of Interest to be addressed in the regulatory submission that will be informed by the computational model. Identify the question, decision or concern that is being addressed using the computational model and other sources of information.
- Define the Context of Use (COU) which is the specific role and scope of the model that addresses the question(s) of interest. It includes –
- a description of what will be modeled.
- how the model outputs will be used to answer the question(s) of interest.
- any additional information that may be included in conjunction with the model to answer the question(s)of interest.
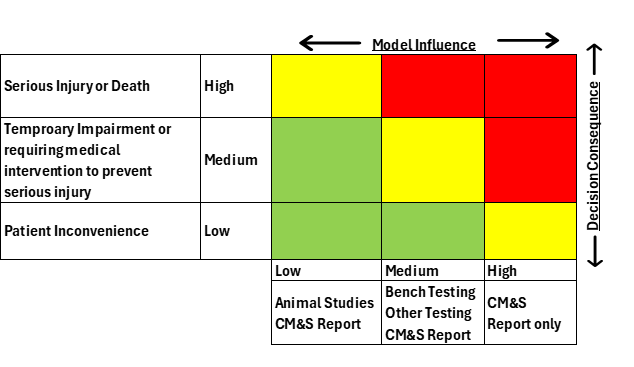
- Determine the model risk, to understand the possibility that the computational model and simulation results may lead to an incorrect decision that would lead to an adverse outcome. Model risk is a combination of model influence and decision consequence. Model influence is the level of contribution of the computational model along with other contributing evidence in addressing the question of interest. Decision consequence is the significance of an adverse outcome resulting from an incorrect decision concerning the question of interest. A possible scheme for assessing model risk considering the combined impact of model influence and decision consequence using a 3X3 grid is shown in Figure 1.
Prospective Planning
The next three steps in the C&MS Medical Device Submissions FDA Guidance document revolve around planning for execution of the model and identifying evidence that may be required for regulatory submissions. These steps are critical to identify if additional input is required, and companies would benefit from FDA feedback through the Q-Submission Program. FDA Guidance “Requests for Feedback and Medical Device Submissions: The Q-Submission Program (June 2023)” provides additional information on the Q-Submission program.
- Identify and categorize the planned credibility evidence that will establish the trustworthiness of the predictive capabilities of the model. This is the expected outcome from the model. Credibility evidence is established by verification, validation and uncertainty quantification (VVUQ).
The guidance provides eight categories of credibility evidence that can characterize the evidence to support a computational model. The eight categories and their definitions are provided in Figure 2.
| Category | Definition | |
|---|---|---|
| 1 | Code verification results | Results showing that a computational model implemented in software is an accurate implementation of the underlying mathematical model |
| 2 | Model calibration evidence | Comparison of model results with the same data used to calibrate model parameters. |
| 3 | Bench test validation results | Validation results using a bench test comparator. May be supported by calculation verification and/or UQ results using the validation conditions. |
| 4 | In vivo validation results | Same as previous category except using in vivo data as the comparator. |
| 5 | Population-based validation results | Comparison of population-level data between model predictions and a clinical data set. No individual-level comparisons are made. |
| 6 | Emergent model behaviour | Evidence showing that the model reproduces phenomena that are known to occur in the system at the specified conditions but were not pre-specified or explicitly modeled by the governing equations. |
| 7 | Model plausibility evidence | Rationale supporting the choice of governing equations, model assumptions, and/or input parameters only. |
| 8 | Calculation verification/ UQ results using COU simulations | Calculation verification and/or UQ results obtained using the COU simulations, that is, the simulations performed to answer the question of interest. |
- Define credibility factors for the proposed credibility evidence by setting prospective credibility goals and establishing a plan to achieve these goals.
- Perform prospective adequacy assessment to ensure that the model can be used for establishing the COU under the risk identified.
Execution, Justification and Reporting
The final three steps in the C&MS Medical Device Submissions FDA Guidance discuss the execution of the proposed study, assessment of data to justify sufficient evidence and development of a report that will be part of the regulatory submission for the FDA.
- Generate the credibility evidence by executing the proposed study(ies) along with analysis of any data from the contributing evidence.
- Determine if the credibility goals were met by assessing the observed outcome of the study(ies) against the credibility goals defined in Step 5. A post-study adequacy assessment will discuss the adequacy of the credibility evidence.
- Prepare a CM&S Credibility Assessment Report that will be a part of the regulatory submission for the FDA.
Conclusion
Using CM&S in the development of medical devices has multiple advantages. It allows manufacturers to test a product before physically developing it by using virtual components and running experiments to confirm the performance. This results in a cheaper development cycle.
CM&S also enables manufacturers to find unexpected problems prior to building, which are then mitigated through design changes. Product development cycle durations may be shorter if there is no need to build and test physical proof-of-concept devices. Additionally, evidence of device performance can be generated without conducting large scale V&Vs by assessing large datasets/population factors.
Medical Device companies are required to properly assess the risk of using CM&S in the development of medical devices to ensure that the overall device safety and performance is not compromised.
The C&MS Medical Device Submissions FDA Guidance summarized in this article provides a nine-step framework for developing and assessing the credibility of CM&S for regulatory submissions. The framework describes defining how the model will be used, performing a risk assessment, planning for the study, execution, justification and generating the report for FDA Submission. CM&S applications will continue to grow and become an important piece in medical device development and play a larger role in documenting evidence of safety and performance in regulatory submissions.
Dhruvitha Krishna is a Quality Assurance and Regulatory Affairs Specialist at StarFish Medical with a M.S degree in Biomedical Engineering. She has worked in manufacturing, new product implementation, software deployment, project management and regulatory areas in medical device companies. Dhruvitha is dedicated to quality, regulatory and continuous process improvement for the manufacturer.
Images: FDA
ELLA is a free AI-powered FDA regulatory research tool that uses advanced natural language processing to extract key insights from FDA databases, guidance documents, and adverse event reports.