
Similarities and Differences between Medical Device 510(k) and CE Marking
This article highlights some of the similarities and differences between the Medical Device 510(k) and CE Marking regulatory pathways and helps harmonise some aspects of an overall regulatory strategy.
Editor’s Note: This article updates and replaces Vincent Crabtree’s 2014 blog on 510(k) and CE Marking (Pt 1 and Pt 2).
FDA has a clear definition of the 510(k) pathway which allows manufacturers to bring their products to market at a faster pace and lower cost compared to FDA’s Premarket Approval (PMA) pathway. The European Union CE marking process, that was originally regarded as straightforward if the device was FDA approved, is no longer a trivial process. Introduction of EU’s MDR (2017/745) and IVDR (2017/746) has led to several manufacturers initiating remediation programs to update their documents for compliance and conducting additional testing or clinical studies.
Executive Summary
The Medical Device Directives (MDD) is in transition to Medical Device Regulations (MDR) in the EU which re-classifies medical devices into Class I, Is, Im, Ir, IIa, IIb, III and IIIc (custom). The device classification guides the requirements for design, testing, verification, validation, clinicals and post market surveillance for the device to be CE Marked and placed on the EU Market.
Medical device 510(k) and CE Marking processes cannot be compared on a one-to-one basis because the 510(k) process applies to a relatively small number of products when compared to applicability of the CE Marking process. Significant changes to devices require some level of monitoring and reporting in both processes and may lead to the need for a new 510(k) or a re-inspection by Notified Bodies.
The 510(k) process has evolved to be straightforward with the eSTAR program and progress tracking functionality. The eSTAR program is currently being piloted for a joint FDA and Health Canada portal, which will make the process more convenient for manufacturers.
(Updated Dec 2024) EUDAMED has three live modules, Actors, Devices, and Certificates & Notified bodies. We are still waiting for the final three modules, later, the new EUDAMED timelines are here with EUDAMED being designated as full function in Q4 2025.
Both 510(k) and CE Marking processes operate under different sets of regulations for the Quality Management System, but ISO 13485:2016 can be employed with additional implementation of FDA and EU MDR clauses. With the proposal to harmonise QSR with ISO 13485:2016, implementing a Quality Management System will become easier for manufacturers. Core design and risk management documentation is required for submission, and supplemental documentation varies based on the market.
Join over 6000 medical device professionals who receive our engineering, regulatory and commercialization insights and tips every month.
Medical Device 510(k) and CE Marking Terminology, equivalence and application
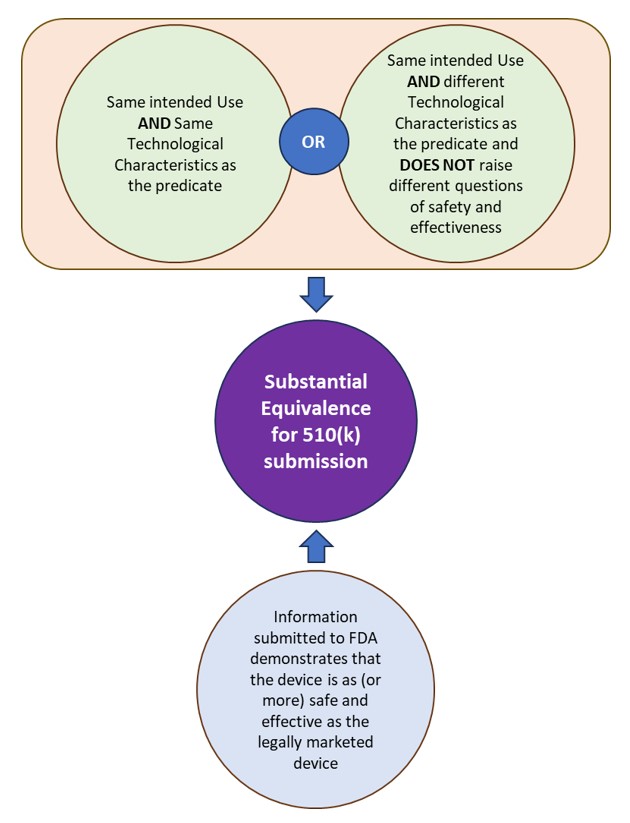
510(k) is a premarket submission made to FDA to demonstrate that the device to be marketed is as safe and effective, i.e. substantially equivalent (SE), as a legally marketed device. The FDA defines the legally marketed device(s) to which equivalence is drawn as the “predicate”. A device is substantially equivalent to a predicate if the subject device:
- has the same intended use as the predicate and has the same technological characteristics or different technological characteristics that do not raise different questions of safety and effectiveness.
- information submitted to FDA demonstrates that the device is as (or more) safe and effective as the legally marketed device.
See Figure 1: Substantial Equivalence under FDA for 510(k) submissions.
510(k) submissions are applicable to Class I, II devices (unless exempt) and Class III devices, if applicable.
Although every country in the European Union, typically referred to as Member States, has a Competent Authority that is responsible for compliance with the device related to CE marking directives, the responsibility of CE marking is delegated to Notified Bodies, to prevent conflict of interest, and harmonise requirements.
To obtain a CE Mark for the device, the manufacturer is required to demonstrate that their device complies with the EU MDR requirements. Typically, Notified Bodies are tasked with reviewing the Device Master Record and associated documents to approve CE Marking for the device. One of the ways of demonstrating compliance is through equivalence. CE Marking, by claiming equivalence under EU MDR, requires additional considerations to demonstrate equivalence. The manufacturer is required to claim equivalence for the following characteristics:
- Technical – conditions of use, specifications and properties, deployment methods (where applicable), principles of operation and critical performance requirements
- Biological – materials or substances in contact with the same human tissues or body fluids, similar kind and duration of contact and release characteristics of substances (including degradation products and leachables)
- Clinical – clinical condition or purpose, severity and stage of disease, site in the body, population, user, critical performance in view of the expected clinical effect for a specific intended purpose.
Demonstrating equivalence under CE Marking requires a lot more effort and sometimes proprietary information that may not be available for attempting to claim equivalence with products belonging to a different manufacturer. Notified Body approval for CE Marking is not required for Class I under EU MDR. Class I devices are self-certified under EU MDR. Class I devices that are provided as sterile (Is), have a measuring function (Im) or are a reusable surgical instrument (Ir) are subject to CE Marking.

Medical Device 510(k) and CE Marking Changes to Device
Devices that undergo design changes may require a new 510(k) submission. A new 510(k) should be submitted in the following cases:
- Changes made with intent to significantly affect safety or effectiveness of a device.
- Major Labeling changes – Adding contraindications, devices re-labelled as re-usable from single use, etc.
- Major Technology, engineering, and performance changes – changes in control mechanism, operating principle, energy type changes, etc.
- Materials changes
- Modifications leading to significant change in the device’s risk profile.
While the list above seems straightforward, there are several scenarios where the lines blur and the device may not require a new 510(k). The FDA guidance Deciding When to Submit a 510(k) for a Change to an Existing Device provides detailed flowcharts that assist with decision making for submitting a new 510(k). A separate guidance discusses when changes to software require a new 510(k).
Under EU MDR, devices undergoing any significant changes in one or more of the following categories should be reporting the change to the Notified Body that certified the device.
- intended purpose
- design or performance specification
- ingredient or material
- sterilization or packaging design with impact on sterilization
- software
The definition of significant change can be ambiguous. MDCG Guidance provides additional information on significant changes under EU MDR. The Notified Body may decide to re-audit the manufacturer’s QMS or technical documentation, as needed.
A good regulatory strategy will comprise of both FDA and EU MDR requirements to ensure that they are both met.
Medical Device 510(k) and CE Marking Processes and Fees
510(k) submissions can be done online using the CDRH portal. The CDRH portal allows manufacturers to send a Medical Device submission using eSTAR, an interactive PDF form. The CDRH portal also provides a progress tracker that displays the submission status. The portal has a few limitations with file size and types but allows the correspondent to mail oversized documents to the CDRH Document Control Center (DCC). Any device that has been granted 510(k) clearance can be placed on the market immediately while awaiting the FDA Quality System (21 CFR 820) inspection, which may take place after the clearance.
Application costs for 510(k) are published on the FDA website under Medical Device User Fee Amendments (MDUFA). The fees vary for standard businesses and small businesses. The fees are updated every fiscal year. Every establishment is required to pay an annual establishment registration fee.
The CE Marking process requires the involvement of Notified Bodies and a qualifying QMS Audit before a device can be evaluated for a CE Mark. Choose your Notified Body based on the right combination of fees, travel arrangements and expertise that works for you.
The EU MDR introduced the concept of a European Database on Medical Devices (EUDAMED) database that is intended to provide a “living picture of the lifecycle of medical devices that are made available in the European Union (EU)”. EUDAMED will be composed of six modules related to: actor registration, unique device identification (UDI) and device registration, Notified Bodies and certificates, clinical investigations and performance studies, vigilance and market surveillance. As of March 2024, EUDAMED has three live modules – economic operators, devices and certificates. The final three modules are scheduled to go online in , the new EUDAMED timelines are EUDAMED being designated as full function in Q4 2025.
In the absence of an online system to manage the submissions for CE Marking, the manufacturer is required to use portals/share folders/other methods set up by Notified Bodies to share the documentation and evidence required for certification. The Notified Body conducts a QMS Audit against the EU MDR Quality Management System requirements (Annex IX) prior to certifying the device for the market. Devices cannot be CE marked and placed on the market without a Notified Body QMS audit and technical documentation review (or a sampling in some cases), unless the device is categorized as Class I.
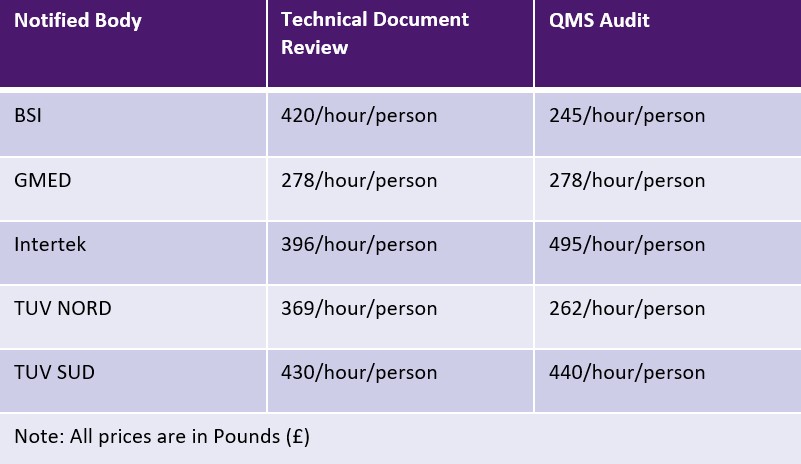
EU MDR allows Notified Bodies to charge either a flat fee or a time-based fee for each activity. However, Notified Bodies are not bound by any limits for the charges. The table below shows a sampling of Notified Bodies and their fees. Additional fees for travel and expertise may be applicable.
Quality Management System for Medical Device 510(k) and CE Marking
Manufacturers are required to do their due diligence and ensure that the appropriate Quality Management System is in place, prior to placing their devices on the market.
The FDA governs the quality management system under the Quality System Regulations (QSR) (21 CFR 820), that applies to manufacturers intending to commercially distribute medical devices. A new rule has been passed to incorporate the ISO 13485:2016 Medical Devices – Quality Management Systems within the QSR in an effort to harmonise the requirements across the board. ISO 13485:2016 is used by many other regulatory authorities around the world as the standard for Quality Management Systems. While the flexibility of 510(k) clearance allows manufacturers to place their device on the market, they may be inspected by the FDA at any time. Hence, it is in the best interest of the manufacturer to ensure a compliant Quality Management System is in place prior to placing the device on market.
With EU MDR, ISO 13485:2016 is not a requirement for CE Marking. The Quality Management System in place should comply with the regulations set forth in Annex IX. Manufacturers must complete an application for assessment of their Quality Management System with the Notified Body. In addition to a Quality Management System audit, Notified Body will assess the technical documentation for the devices against the Quality Management System to ensure that all the requirements are met before certification.
To harmonise the Quality Management System, the best practice is implementing an ISO 13485:2016 and Medical Device Single Audit Program (MDSAP) compliant system with additional compliance requirements for FDA, EU MDR, Health Canada or other countries where the manufacturer intends to do business.
Medical Device 510(k) and CE Marking Documentation
The FDA and EU MDR require a controlled design process for devices.
510(k) submission requires a complete Design History File (DHF) detailing the device system requirements, architecture, specifications, verification and validation and documentation of the risk management activities. The 510(k) eSTAR profile allows manufacturers to attach the related document to each section of the application. FDA provides an Acceptance Checklist to guide the manufacturer with the 510(k) process and ensure that the right documentation is in place.
For EU MDR, a Technical File contains the DHF documents and also requires the manufacturer to provide the following additional checklists:
- General Safety and Performance Requirements (GSPR) under Annex I, previously known as Essential Requirements Checklist under MDD
- Standard Technical Documentation (SteD) under Annex II
- Post-market surveillance (PMS) under Annex III
A high-level technical document typically references the various DHF documents and the checklists.
In both cases, a full risk management file compliant with the ISO 14971 is required.
References:
- Premarket Notification 510(k) | FDA
- The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications [510(k)] (fda.gov)
- Deciding When to Submit a 510(k) for a Change to an Existing Device – Guidance for Industry and Food and Drug Administration Staff (fda.gov)
- Deciding When to Submit a 510(k) for a Software Change to an Existing Device – Draft Guidance for Industry and Food and Drug Administration Staff (fda.gov)
- MDCG 2019-15, Guidance Notes for Manufacturers of Class I Medical Devices
- Send and Track Medical Device Premarket Submissions Online: CDRH Portal | FDA
- EUDAMED database – EUDAMED (europa.eu)
- Medical Device User Fee Amendments (MDUFA) | FDA
- MDCG 2023-2, List of Standard Fees
- Quality System (QS) Regulation/Medical Device Good Manufacturing Practices | FDA
- Acceptance Checklists for 510(k)s | FDA
- eSTAR Program | FDA
- FDA Health Canada eSTAR (starfishmedical.com)
Images: Adobe Stock & StarFish Medical
Dhruvitha Krishna is a Quality Assurance and Regulatory Affairs Specialist at StarFish Medical with a M.S degree in Biomedical Engineering. She has worked in manufacturing, new product implementation, software deployment, project management and regulatory areas in medical device companies. Dhruvitha is dedicated to quality, regulatory and continuous process improvement for the manufacturer.