FDA’s Premarket Evaluation Team “hit list” for human factors guidance
Did you know that this year FDA’s Human Factors Premarket Evaluation Team have a priority review hit-list for medical devices? Are you on it, are you ready, and what does it mean? This blog will help you understand what it means for you and your device.
A Little Premarket Evaluation History and Context
On February 3, 2016, FDA Human Factors Premarket Evaluation Team formally released its guidance ‘Applying Human Factors and Usability Engineering to Medical Devices’, superseding the ‘Medical Device Use-Safety: Incorporating Human Factors Engineering into Risk Management’ released 16 years previously. The formal release of this guidance was a huge milestone in forwarding and solidifying the human factors and usability engineering process in medical devices development.
On the same day, a new draft guidance was also released titled “List of Highest Priority Devices for Human Factors Review”. FDA decided which device types they believe have clear potential for serious harm resulting from use error. With this draft guidance they have shown their hand on what they will be looking at first and how they will be testing their application guidance.
FDA’s premarket evaluation team issued the Priority List Guidance to inform medical device manufacturers which 60 device types ‘should’ have human factors data included in premarket submissions in accordance with the new Application of Human Factors Guidance. It should be noted that use of the ‘should’ impresses that the priority list is not yet legally enforceable. It is, however, strongly suggested as a recommendation. My view is that you should develop to the new application guidance as part of best practice, and more so, because by the time you reach your submission date your device may be on the list and the word ‘should’ could have turned to ‘shall’.
For those Devices on the Premarket Evaluation list
In the words of FDA, any premarket submission for the device types listed should include either a human factors test report and data as described in ‘Applying Human Factors and Usability Engineering to Medical Devices’, or should provide a detailed rationale that supports the conclusion that human factors data are not necessary.
What human factors data are they looking for in a premarket submission?
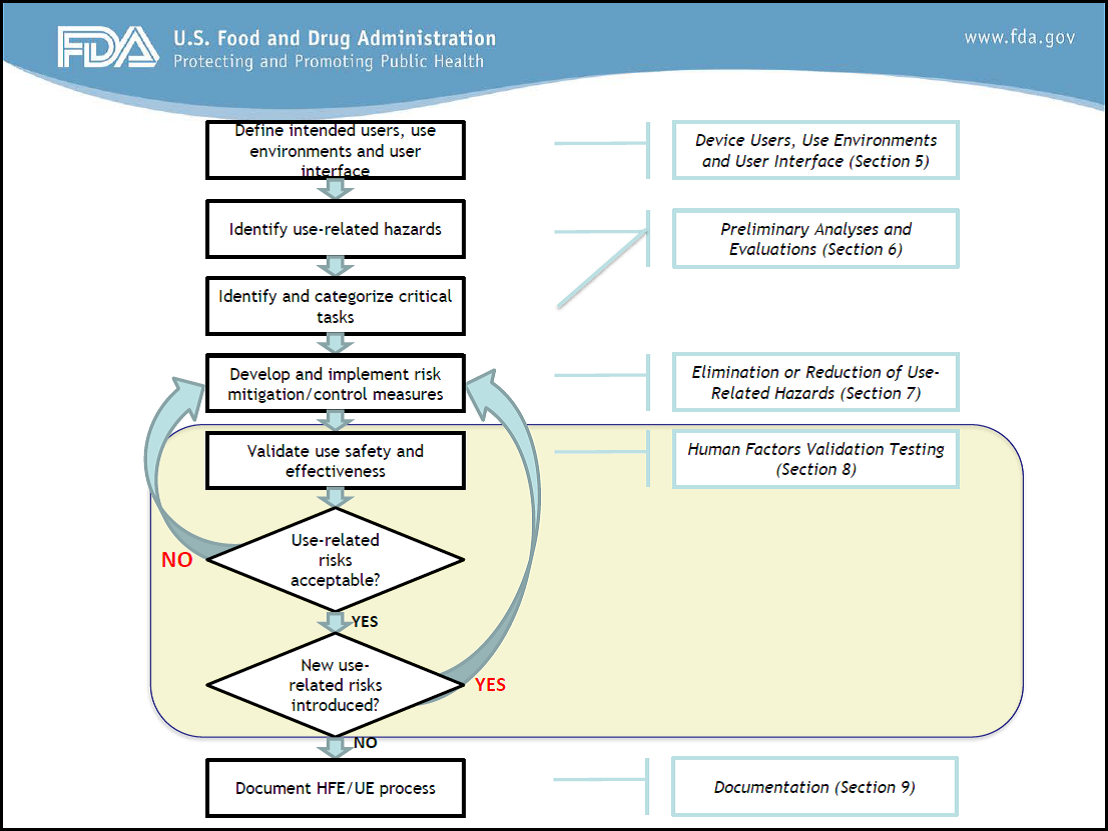
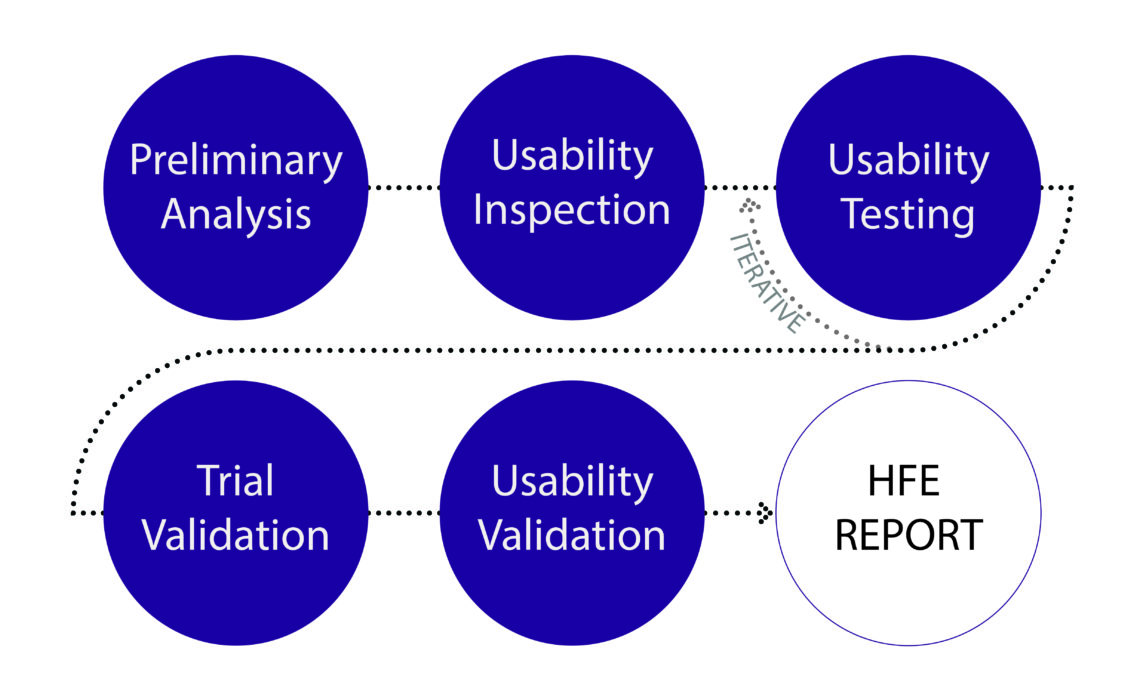
In a nut shell, FDA want manufacturers to provide a report that summarizes the human factors and usability engineering processes they have followed from the initial development, including any preliminary analyses, formative evaluations, human factors validation testing, results and conclusions. You can find more information on this in Appendix A of ‘Applying Human Factors and Usability Engineering to Medical Devices’. In a future blog we will cover the importance of a robust submission report, with some of the potential pitfalls and opportunities.
So what rational would support that human factors data is not necessary if you’re on the Premarket Evaluation list? FDA has stated that your rational should be based on an analysis of risk and the results of that analysis would definitively show that the severity of the potential harm resulting from use error is not serious. It would be required to prove this to a minimum context of users, user tasks, user interface, or use environments from those of the predicates. In my view proving the exception would be as much of a challenge in many cases as conducting a lean Human Factors and Usability report and proving that your device is safe and effective within the same contexts. Of course there are always exceptions and that is for you to decide.
For those Devices not on the Premarket Evaluation list
This is a priority list and we must stress that its wording should not be viewed as an exception list. FDA have clearly outlined within the document that the only exceptions to reporting in accordance with the application guidance will be in cases where the analysis of risk indicates that users performing tasks incorrectly or failing to perform tasks could not result in serious harm. In general, this can only be concluded through some sort of early usability evaluations.
The exemption outlined is tempered within the same document stating that each device will be considered on a case by case basis and may require human factors reporting in accordance with the new application guidance where the submission type is Premarket Application PMA or De Novo Petition, where user interface modification (new or different) are implemented, change of intended users, recalls, adverse events, problem reports and device modifications.
The exemption is further tempered within the same document as FDA outline that human factors data be included in premarket submissions for device types through product specific guidance documents, special controls guidance or guideline documents, or special controls contained in medical device classification regulations. Dangerous words for those seeking exemptions.
The Premarket Evaluation List:
Ventricular assist devices (e.g., DSQ, PCK)
Ablation generators (associated with ablation systems, e.g., LPB, OAD, OAE, OCM, OCL)
Anesthesia machines (e.g., BSZ)
Artificial pancreas systems (e.g., OZO, OZP, OZQ)
Auto injectors (when CDRH is lead Center; e.g., KZE, KZH, NSC)
Automated external defibrillators
Duodenoscopes (on the reprocessing; e.g., FDT) with elevator channels
Gastroenterology-urology endoscopic ultrasound systems (on the reprocessing; e.g., ODG) with elevator channels
Hemodialysis and peritoneal dialysis systems (e.g., FKP, FKT, FKX, KDI, KPF ODX, ONW)
Implanted infusion pumps (e.g., LKK, MDY)
Infusion pumps (e.g., FRN, LZH, MEA, MRZ )
Insulin delivery systems (e.g., LZG, OPP)
Negative-pressure wound therapy (e.g., OKO, OMP) intended for home use
Robotic catheter manipulation systems (e.g., DXX)
Robotic surgery devices (e.g., NAY)
Ventilators (e.g., CBK, NOU, ONZ)

Last thoughts
The premarket evaluation team will be rapidly broadening their focus to a large range of devices. Although your device may not yet be on the priority hit list, by the time you submit your 510 (k) or PMA, it may well be. My recommendation is to assume you are on the list and to work to the full new guidance. It is best practice and will render you a great device that users will love and regulatory bodies will accept. That’s what we have been doing since our Human Factors group was established over 10 years ago and it has worked great for our clients.
Reference: List of Highest Priority Devices for Human Factors Review, Draft Guidance for Industry and Food and Drug Administration Staff. Document issued on February 3, 2016.
James Jackson is a former StarFish Medical industrial designer and project manager. He utilizes a multidisciplinary approach to managing client projects. This is his first blog for StarFish. He previously wrote on FDA Expectations for Human Factors for MedTech Intelligence.
Images: StarFish Medical
Your Roadmap for Premarket Submissions: FDA Guidance on Non-Clinical Bench Performance Testing Information