Planning, Input and Architecture Tips for Medical Device Design Documentation
I sometimes find myself in discussion with Entrepreneurs who have developed a novel prototype Medical Device and are ready for market launch, but the device documentation available is an electronic schematic, source code and a Google Sketchup of the enclosure. As a Regulatory Advisor, I explain they must follow a documented medical device development process; effectively ‘Start again’.
As you could imagine, this news goes down like a lead balloon, as no one reacts well to their baby being called ugly. In this blog, I will discuss 3 medical device design documentation tips that anyone developing early stage medical device should adopt. They make the transition from prototype to product less effort, reducing development timescales and cost.
Prototypes vs. Products
I worked as a scientist at a University in England where we performed Healthcare based research and developed innovative medical technology. We believed the technology we were developing was inches away from being real products. So, when I first went to a commercial medical device development consultancy after raising money I was offended when the Business development person told me I didn’t have enough!
It is important to remember a prototype is something that is not going to be marketed or sold, and a product is something we will sell. Formal design controls are not required for prototypes. We only have a legally sellable medical device product after going through a product realization process.
The more rigour applied at an early stage, the easier the transition into an actual product will be, reducing development costs and timescales.
Documentation Tips
Reviewing the FDA 21 CFR 820 QSR regulations would be time well spent for anyone developing Healthcare technology, as it sets the required groundwork.
Design Planning
The most important of my medical device design documentation tips is to plan the design and development, even though you may only be developing a prototype. In addition to project planning activities (such as a Gantt chart), the Design Plan should also contain at least the following:-
- The ‘Indications for Use’ statement – this helps define the risk for the device, and succinctly defines what it is you actually want your device to do, which is essential for ensuring all members of your team are on the same page. The simpler and clearer this statement, the better. For example:
The XXYYZZ Surgery System is indicated for use in general, plastic and reconstructive surgical procedures to cut and coagulate soft tissue.
- Applicable Standards – there are Harmonized standards and FDA guidance documents for many different devices. These range from performance standards for ultrasound, ECG, pulse oximeters, blood glucose meters etc. to general standards for EMC, Basic Safety and Essential performance and others, see below.
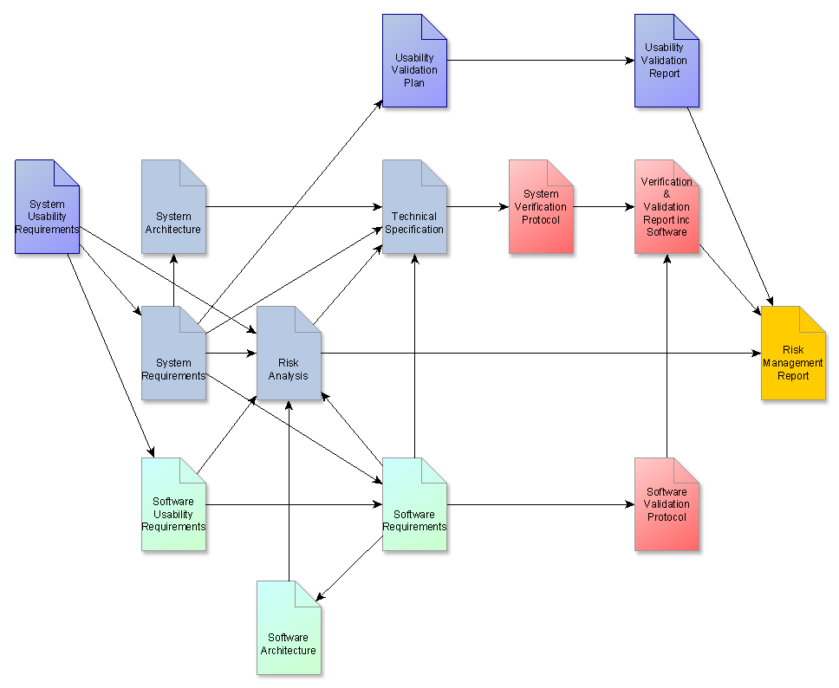
- Document Map – a clear Document Map really helps a multidisciplinary project, which shows the relationship between each document, like the one below:
The document map clearly indicates the design input documents, the technical specs, verification and validation activities, and risk activities.
- Design Reviews – This is probably the biggest departure for those not involved in formal design processes; one typically sit at a computer and fiddle about using their favourite software package (IDE & C compiler, 3D modeling software, Schematic Capture etc) to develop a design which they are happy with, with a rough idea of what it should do. However, a design review compares a set of formal Requirements against the resulting design which has been generated, and this is done by Peers. The intent is to catch things the original designer did not, and confirm the resulting design is fit for purpose. All major components and assemblies should undergo design review (Software modules, circuit sub-assemblies, mechanical assemblies etc), and the Design Plan should state who will attend and when these will happen, with a gated approach.
Design Input Requirements
Design input documents are commonly missing. Based on the Indication for Use, the input documents describe Requirements which will achieve that indication, expanded into Usability, System and Technical Requirements. For example, the Usability Requirements may say that a coagulating scalpel is required with a diameter less than 1 inch. From this, the System requirements may state that an RF heater is required which can cauterize 1g of tissue per second. The Technical Specs will elaborate further with a line item that calls for the system to emit 20Watts of RF power at 4MHz. Each Requirement line item is testable, numbered and has a source reference where applicable, the Technical Spec references the System Requirements, which may also reference the Usability Requirements.
Architecture Document
Requirements Documents for early stage work are often confused, containing soft requirements, technical specifications and untestable statement like ‘Must be easy to use’. The medical device design documentation tip here is to a) improve untestable requirements so they can be tested or remove them, and b) relegate ‘Soft’ requirements to the architecture document, as these are really guidance to the designers and should not affect safety and performance. Typical soft requirements are logos, colour schemes, design aesthetic etc. The Architecture document illustrates the relationship between the various internal elements, and can also be used to define the interface between subsystems if multiple Architecture Documents are required, which also helps with team communication.
Conclusion
With a good document base, it is much more straightforward to take prototypes to a product, as we can start product realization much more quickly without requiring a long ‘Product Definition’ Phase. My next medical device design documentation tips blog will cover Medical Device Design Documentation Risk Management, Verification & Validation (V&V), and Standards tips.
Vincent Crabtree, PhD is a Regulatory Advisor & Project Manager at StarFish medical, and would be delighted to hear comments and constructive criticism on this article.
Images: StarFish Medical