FDA Final Guidance on Remanufacturing of Medical Devices: Key Takeaways and Implications
This blog breaks down the key aspects of the FDA’s remanufacturing guidance document, its potential regulatory impacts, and best practices for compliance.
In May 2024, the FDA released an updated guidance document to help manufacturers, third-party service providers, and healthcare facilities determine whether their modifications fall under the category of remanufacturing. This guidance enables these entities to continue ensuring patient safety, regulatory compliance, and the effectiveness of remanufactured medical devices.
Scope
The FDA’s guidance on remanufacturing aims to provide clarity on regulatory requirements for entities involved in modifying medical devices. The guidance distinguishes between servicing and remanufacturing, emphasizing the need for proper classification to ensure compliance with safety and performance standards. This guidance applies to all reusable medical devices, including software-based and electronic products that meet the legal definition of a device. However, it does not cover single-use devices that are reprocessed.
FDA defines Remanufacturingas the processing, conditioning, renovating, repackaging, restoring, or any other act done to a finished device that significantly changes the finished device’s performance or safety specifications, or intended use [1] and Servicing as the repair and/or preventive or routine maintenance of one or more parts in a finished device, after distribution, for purposes of returning it to the safety and performance specifications established by the OEM and to meet its original intended use [2]
Guiding Principles
To help manufacturers and service providers assess whether an activity qualifies as remanufacturing, the FDA outlines several guiding principles. These principles ensure that modifications to medical devices do not compromise safety, performance, or regulatory compliance.
- Consider the Cumulative Effect of Multiple Changes – Even if individual modifications seem minor, their combined impact can significantly alter the device’s function. A holistic review ensures that small adjustments do not collectively result in remanufacturing.
- Perform a Risk-Based Assessment – Using ISO 14971 Risk Analysis or similar frameworks, entities should evaluate whether changes introduce new risks or significantly modify existing ones. A well-documented risk analysis supports regulatory decision-making.
- Maintain Comprehensive Documentation – Suggested throughout the guidance document, keeping proper records for every modification, including risk assessments, testing results, and justification for why a change is or isn’t considered remanufacturing, is the key to compliance. The FDA expects this level of documentation for regulatory audits and inspections.
The guidance document recommends utilizing these principles to assess the impacts of the changes being made by the manufacturer or service provider. Throughout the guidance document, the reader is suggested to document approaches pursued, test data collected, and other relevant information gathered, where applicable. By following these principles, manufacturers and third-party service providers can make informed decisions about device modifications while ensuring compliance with FDA regulations.
How to determine if the activities are remanufacturing
Determining whether an activity qualifies as remanufacturing requires a structured evaluation of the modifications made to a medical device. The guidance document provides a decision-making framework, including specific criteria and a step-by-step assessment process to guide manufacturers and service providers.
- Does the Modification Change the Device’s Intended Use? – If an activity modifies the device’s function, application, or patient population, it is likely remanufacturing
- Does the Change Significantly Impact Performance or Safety? – Changes to critical components, software, or materials that alter the device’s operation, durability, or sterility are strong indicators of remanufacturing.
- Are There New or Modified Risks? – Any modification that introduces new hazards or alters existing risk levels must be carefully assessed.
The guidance document also provides a flowchart to help the manufacturers and service providers determine if their activities qualify as remanufacturing.
Figure 1: Flowchart to help determine whether activities performed are likely to qualify as remanufacturing. Flowchart Source: FDA [2]
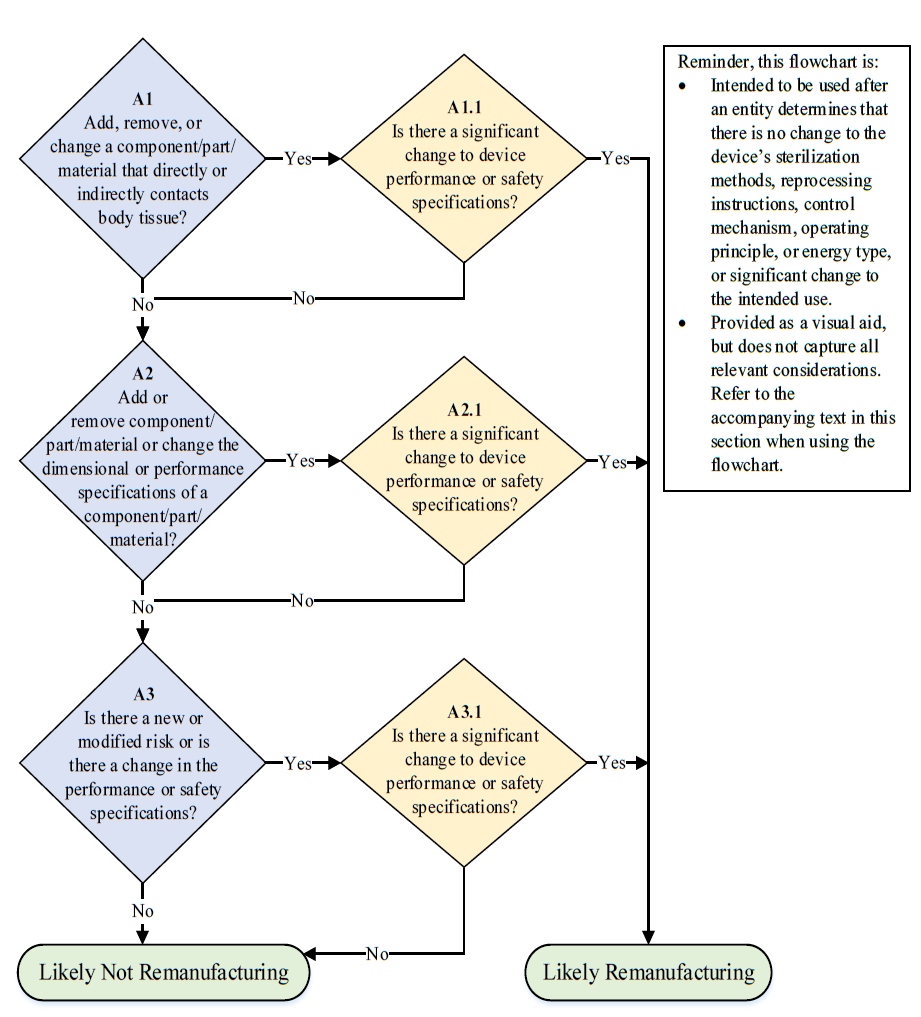
The guidance document also provides a flowchart to help the manufacturers and service providers determine if their activities qualify as remanufacturing.
Regulatory Requirements and Considerations for Remanufacturers
The FDA considers remanufacturers to fall under the same regulations as manufacturers, and entities that are remanufacturing devices, are subject to the same regulatory requirements as the original manufacturer of the device.
The FDA outlines a few key areas of compliance that must be adhered to for remanufacturing:
Establishment Registration and Medical Device Listing
Premarket Authorization (510(k) or PMA Submission)
Reports of Corrections and Removals
Quality System (QS) Regulation Compliance
Labeling and Unique Device Identification (UDI) Requirements
Remanufacturers must register their facilities with the FDA and list their remanufactured devices separately from the OEM’s device listing.
If a modification significantly alters a device’s safety, performance, or intended use, the remanufacturer must obtain premarket authorization.
Under 21 CFR Part 806, remanufacturers must report any corrections or removals of devices that are made to reduce health risks posed by the device or to address FDA violations
Remanufacturers must establish a Quality Management System (QMS) that meets the FDA’s Quality System Regulation (21 CFR Part 820).
If a modification changes the device’s function or safety, the remanufacturer must update the labeling to reflect the changes.
Conclusion
The remanufacturing of medical devices is a highly regulated process that requires strict adherence to FDA guidelines to ensure patient safety and device effectiveness. Misinterpreting remanufacturing as routine servicing can lead to compliance risks, regulatory penalties, and potential harm to patients.
The FDA’s guidance provides a clear framework for distinguishing between servicing and remanufacturing. By following the guiding principles, using risk-based assessments, and maintaining comprehensive documentation, manufacturers and service providers can make informed decisions about device modifications. By fostering a culture of compliance, transparency, and safety, companies can contribute to the long-term reliability of reusable medical devices while supporting advancements in healthcare.
For more details and examples, refer to the FDA’s official guidance on Remanufacturing of Medical Devices [2].
Soumyaa Varshni is a Quality Engineer at StarFish Medical. She received her Master’s in Biomedical Engineering from Cornell University in 2017, with a focus in manufacturing medical devices.
Images: Adobe Stock
References
[1] https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-820/subpart-A/section-820.3
When it comes to risk, the first thing to consider is the ISO 14971 standard. MDR and IVDR changes bring a new alignment in terms of risk.
Related Resources

Nick Allan and Nigel Syrotuck explain exactly how aminoglycoside antibiotics work and why they’re so effective at killing bacteria.

Graphical mind maps created in online whiteboards offer a low-barrier, highly collaborative approach to early risk analysis in medical device development.

Early phase concept development is a weird part of a project lifecycle. It is often the most exciting phase, because the team is exploring possibilities, generating new ideas, and turning a fuzzy opportunity into something real.

Clinical prototypes must not only function as intended, but also be manufactured, documented, and supported in a way that satisfies regulatory expectations and clinical realities.
Biocompatibility is a significant aspect of contact, but what about sterility? And if a device does not need to be sterile, then how clean does it need to be?